From viral bio-distribution studies done for this virus in control conditions or in presence of TSA. We further tested the possibility that the combination of B18R-deleted vaccinia virus and TSA could be effective in a human xenograft tumour model. Immunocompromised mice with palpable HCT-116 colon cancer tumours were treated with TSA and a luciferase-expressing B18R/TK-deleted virus and IVIS imaging was used to examine the growth of the virus in tumour bearing mice. Treatment with TSA resulted in Z-VAD-FMK increased virus-associated luciferase activity within HCT-116 tumours when compared to treatment with vaccinia virus alone. The low virus signal in the lungs after 48 h is consistent with the biodistribution data; however this signal is gone by 4 days. Importantly the signal in the lungs is not enhanced by TSA treatment whereas the signal is greatly enhanced in the tumour. Consistent with this observation and the results obtained in the lung metastasis model, mice treated with the combination of TSA and TK/ B18R-deleted WR had delayed tumor progression and demonstrated increased survival versus mice treated with either agent alone. Trichostatin A was one of the first HDAC inhibitors to be discovered and although its anti-cancer properties are well documented, its sub-optimal in vivo stability has made it less attractive for use as a chronically administered anti-cancer drug. A considerable effort in the HDI field has led to the development of more stable TSA derivatives such as Vorinistat H, which was recently approved for limited applications like treatment of CTCL. In this study we found that SAHA was significantly less effective at augmenting vaccinia virus spread in vitro than TSA. In this context, we also found that TSA interacted synergistically with vaccinia virus, leading to better cell killing. Theoretically, this synergistic interaction predicts that the effective dose used for each therapeutic in vivo could be reduced while retaining efficient anti-tumour activity. However, since OVs need to overcome numerous physiological barriers in order to reach tumors, it is likely that TSA/Vaccinia combination therapy would be best used as a means to increase efficacy as opposed to dose reduction. Nonetheless, this suggests that the relatively short half-life of TSA in vivo may not be a concern for the therapeutic application described here in light of the relative potency of its vaccinia-enhancing effect. Oncolytic virus therapy is an acute treatment with curative intent. Indeed the activity of OVs involves not only SAR131675 1433953-83-3 replication in, and destruction of tumour cells but also the recruitment of host immune cells to the tumour bed leading to the initiation of antitumour immunity. It is known that HDIs can impact the patient��s immune cells and thus a fast acting, virus enhancing, compound that is rapidly cleared once an infection is established may be preferred. Furthermore the short half-life of TSA allows for better control over the OV dose should treatment need to be stopped abruptly. Given that our data shows anti-tumoral activity of several different VV strains can be enhanced by TSA in vivo the clinical application of TSA may need to be re-visited. This is of considerable interest since VV strains such as JX-594 and JX-929 are currently undergoing Phase I/II clinical trials. The effect of TSA on the IFN response is well documented and the enhancing effect of HDIs such as TSA on IFNsensitive strains including VSV and HSV has  been previously reported. It is therefore not surprising that TSA can increase the activity of B18R-deleted VV strains.
been previously reported. It is therefore not surprising that TSA can increase the activity of B18R-deleted VV strains.
Author: screening library
Without killing the pathogen bacteria may cause less selective pressure for the generation
Medicinal chemistry usually adopts weak positively charged groups to increase the membrane permeability of candidate drugs that easily pass through the porins, as in the case of BZD and other positively charged derivatives. In this case, however, the option of a porin mutation is available and bacteria might develop a rapid resistance to these drugs. This resistance mechanism can be overcome by employing molecules that permeate directly through  the bacterial membrane, as BZB derivatives. Unfortunately, however, membrane permeation can be slow and this decreases the antibacterial activity potential. Here we provide information on the structural determinants of BZB permeation through the membrane by molecular simulations. Our calculations show that a water-filled channel favors the membrane translocation. These observations could be used for chemical modifiMasitinib cations of BZB to obtain compounds with improved membrane permeability. Several virulence regulator factors, such as two-component signal systems, quorum sensing systems, type III secretion systems, and the assembly of adhesive organelles, have been recognized as interesting targets to reduce bacterial infection. Bacterial two-component systems have gained increasing interest as novel antibacterial targets because these systems are required for virulence of pathogenic microorganisms. PhoQ/PhoP is a two-component system that governs virulence, monitors the extracellular Mg2+, and regulates several cellular activities in many gram-negative species. The system also helps bacteria resist antibiotic peptides by regulating lipid A. Bivalent cations and antibiotic peptides can competitively bind to the acidic structural domain on the cytoplasmic surface of PhoQ. PhoQ is an attractive target for an antibiotic because it is absent in mammals. In this study, we have explored the possibility of using the PhoQ as a potential target by performing a screen for inhibitors. After constructing a 3D model of the PhoQ HK domain of Sf301, 64 compounds were selected as inhibitor candidates based on their molecular diversity, shape complementarities, and potential for forming hydrogen bonds in the binding pocket of PhoQ. To confirm the interaction of the compounds and PhoQ, a prokaryotic expression plasmid containing the Sf301 PhoQ intracellular domain which contains HK domain was constructed, because the main biology activity of PhoQ is depends on its HK domain. To confirm whether these inhibitor candidates targeted the PhoQ HK domain, enzymatic activities of PhoQ were determined in the presence or absence of four compounds. The enzymatic activity of SF-PhoQc was measured using both a Pyrophosphate Reagent and a Luminescent Kinase Assay. The Pyrophosphate Reagent can reflect the Nutlin-3 Mdm2 inhibitor reaction of HK and ATP at real time, but not sensitive. The Luminescent Kinase Assay is more sensitive than Pyrophosphate Reagent for kinase reaction but cannot reflect the reaction of HK and ATP at real time. Therefore, in the present study we used two assays to confirm the results. The different IC50 values of potential PhoQ inhibitors 1 and 3 determined by the two assays may be the sensitivity difference between the two assays. By using cell invasion assays, the features of cell invasion process including penetration into epithelial cells and spreading to adjacent cells were tested. These results indicate that inhibition of microbial virulence without inhibiting their growth may be a promising strategy. In contrast, currently available antibiotics either kill bacteria or prevent their growth.
the bacterial membrane, as BZB derivatives. Unfortunately, however, membrane permeation can be slow and this decreases the antibacterial activity potential. Here we provide information on the structural determinants of BZB permeation through the membrane by molecular simulations. Our calculations show that a water-filled channel favors the membrane translocation. These observations could be used for chemical modifiMasitinib cations of BZB to obtain compounds with improved membrane permeability. Several virulence regulator factors, such as two-component signal systems, quorum sensing systems, type III secretion systems, and the assembly of adhesive organelles, have been recognized as interesting targets to reduce bacterial infection. Bacterial two-component systems have gained increasing interest as novel antibacterial targets because these systems are required for virulence of pathogenic microorganisms. PhoQ/PhoP is a two-component system that governs virulence, monitors the extracellular Mg2+, and regulates several cellular activities in many gram-negative species. The system also helps bacteria resist antibiotic peptides by regulating lipid A. Bivalent cations and antibiotic peptides can competitively bind to the acidic structural domain on the cytoplasmic surface of PhoQ. PhoQ is an attractive target for an antibiotic because it is absent in mammals. In this study, we have explored the possibility of using the PhoQ as a potential target by performing a screen for inhibitors. After constructing a 3D model of the PhoQ HK domain of Sf301, 64 compounds were selected as inhibitor candidates based on their molecular diversity, shape complementarities, and potential for forming hydrogen bonds in the binding pocket of PhoQ. To confirm the interaction of the compounds and PhoQ, a prokaryotic expression plasmid containing the Sf301 PhoQ intracellular domain which contains HK domain was constructed, because the main biology activity of PhoQ is depends on its HK domain. To confirm whether these inhibitor candidates targeted the PhoQ HK domain, enzymatic activities of PhoQ were determined in the presence or absence of four compounds. The enzymatic activity of SF-PhoQc was measured using both a Pyrophosphate Reagent and a Luminescent Kinase Assay. The Pyrophosphate Reagent can reflect the Nutlin-3 Mdm2 inhibitor reaction of HK and ATP at real time, but not sensitive. The Luminescent Kinase Assay is more sensitive than Pyrophosphate Reagent for kinase reaction but cannot reflect the reaction of HK and ATP at real time. Therefore, in the present study we used two assays to confirm the results. The different IC50 values of potential PhoQ inhibitors 1 and 3 determined by the two assays may be the sensitivity difference between the two assays. By using cell invasion assays, the features of cell invasion process including penetration into epithelial cells and spreading to adjacent cells were tested. These results indicate that inhibition of microbial virulence without inhibiting their growth may be a promising strategy. In contrast, currently available antibiotics either kill bacteria or prevent their growth.
All of the selected inhibitors dependent inhibition of APE1 in the gel-based assay
BER Y-27632 dihydrochloride inhibitors or activators would provide novel resources, not only for basic science purposes, but for the potential development of high affinity targeted, therapeutics that may improve the efficacy of treatment paradigms by promoting selective sensitization of diseased cells or increasing the protection of normal cells, respectively. Small molecule protein modulators are viewed as the perfect complement to frequently-used techniques such as RNAi and gene knockout. In addition, the scientific and medical communities have become increasingly interested in the design of small molecule inhibitors of DNA repair, with the potential of improving the therapeutic efficacy of clinical DNA-interactive drugs. While there have been reports of small molecules directed at APE1, the utility of these inhibitors has been brought into doubt given the inability to reproduce their effectiveness or the failure of the compounds to elicit a cellular consequence. It is anticipated that the reconfigured APE1 assay described here will serve as a useful guide to future investigations aimed at screening other nucleic acid processing enzymes. The concentration-response screen of the LOPAC collection yielded a number of previously-unreported APE1 inhibitors. The most potent hit was ATA, which inactivated the enzyme consistently in the low nanomolar range. While very effective, ATA has been noted to exist as a stable radical homopolymer of varying length and to act as a strong inhibitor of a large number of DNA- and RNA-processing enzymes. As a means of interrogating the primary screening hits, and to gain further insight into their mechanism of action, we employed a fluorescent dye-displacement assay, substituting the frequently-used ethidium bromide with the more sensitive and robust reporter ThO. In a screen of the LOPAC collection against a ThO-substrate DNA complex, all annotated fluorescent DNA intercalators within the library, e.g. idarubicin, doxorubicin and distamycin, displayed strong displacement activity. Furthermore, the non-fluorescent APE1 screening hits WB 64 and mitoxantrone exhibited dye-displacement IC50 values similar to or better than those displayed in the APE1 enzymatic incision assay. This behavior is reminiscent of an indirect, non-competitive DNA binding effect and is consistent with the multiple fused ring Epoxomicin systems, which have a tendency for DNA intercalation, featured in both molecules. On the other hand, APE1 inhibitor molecules that lacked obvious DNA-binding features, such as thiolactomycin and Tyrphostin AG 538, yielded weak or no displacement activity. These findings support the ThO displacement assay as a convenient counterscreen to exclude DNA binders from further consideration, and the complete results with the LOPAC1280 are available in the corresponding PubChem deposition. To further probe the selectivity of the inhibitors identified in the APE1 qHTS, we tested the LOPAC collection against E. coli EndoIV. AP endonucleases are sub-classified into two major superfamilies based on homology to either E. coli exonuclease III or E. coli EndoIV. While members of the two superfamilies exhibit similar biochemical properties, such as AP endonuclease activity, there exists no sequence or structural homology between the different superfamily constituents. Among the actives shared between the two endonucleases were the nonspecific DNA binders mitoxantrone and WB 64, whereas compounds with selectivity to APE1 included thiolactomycin, 6hydroxy-DL-DOPA, methyl 3,4-dephostatin, myricetin and Reactive Blue 2. The fact that the EndoIV screen did not identify either potent or EndoIV-selective hits may suggest an active site for this enzyme that is hard to access. Our ongoing studies of inhibitors for E. coli EndoIV are outside the scope of the present investigation and will  be the subject of a separate report. Finally, we tested the top screening hits in a standard radiotracer incision assay.
be the subject of a separate report. Finally, we tested the top screening hits in a standard radiotracer incision assay.
Antitubulin activity is likely to be responsible for selective cytotoxicity against tumorigenic cell lines
Subsequent analysis of the scientific literature revealed that many of our compounds do indeed inhibit polymerization of Carfilzomib tubulin in vitro. Compound 384634 has been synthesized and has shown to demonstrate antitublin activity in a tubulin polymerization assay. Likewise, isosteres of compound 385177, 5468780 and 5468781 potently inhibit tubulin polymerization. It is highly plausible that compound 379512 is an antitubulin agent as well, because a number of compounds containing the 2-phenylquinolone ring structure have been synthesized and exhibit tubulin polymerization. Compound 5388755 is almost structurally identical to Combretastatin A-4, which is a very potent antitubulin agent. COMPARE analysis was performed to further characterize the mechanism of action of the compounds. In COMPARE, a correlation coefficient of 0.6 is generally taken to indicate evidence for similar mechanisms of action between the tested and reference compounds. The higher the correlation coefficient, the more likely it is that the compounds share the same intracellular target. The correlation coefficient of the COMPARE computations for the eight most potent compounds and the antimitotic standard anticancer PD 0332991 agents reveals several compounds showing high correlations with microtubule inhibitors colchicine, maytansine, vinblastine and vincristine. None of these compounds show similarity to any of the agents from other mechanistic classes such as topoisomerase inhibitors, alkylating agents and DNA/RNA antimetabolites. None of the compounds exhibit strong correlation with taxol, which is an antimitotic agent that acts by stabilizing microtubules. In order to identify the role of antitubulin activity in generating selective cytotoxicity, we identified twelve additional DTP compounds that are structurally related to some of the nine compounds we identified in our correlation analysis but that lack antitubulin activity. If antitubulin activity confers selective cytotoxicity, these compounds with no antitubulin activity should demonstrate no selective cytotoxicity. The scatterplot comparing the association between cytotoxicity and take-rate for these twelve compounds indicates that none of these compounds show selective cytotoxicity, and they are largely inactive in the cell growth inhibition assay. By data mining the DTP archive, we are able to identify compounds that are preferentially toxic  against the most tumorigenic of the NCI60 cell lines, based on the take rate of the cell lines in a mouse xenograft model. We also established that the activity of these compounds was not correlated to the expression of cell surface stem cell markers reported in the literature. Nevertheless, tumorigenic potential is the most important functional relationship between the most aggressive tumor cells and in vitro model for drug screening. Therefore, the anticancer agents identified based on their activity against the most tumorigenic cell lines may be considered as candidate anticancer agents that are specifically directed against subpopulations of cancer cells that drive the growth of tumors. One of these agents has been found to inhibit microtubule polymerization. Likewise, isosteres of three of our agents have also been shown to inhibit microtubule polymerization, suggesting a single mechanism of action. Interestingly, Compound 5388755 is structurally related to the potent antitubulin agent Combretastatin A-4. It is also possible that compound 379512 acts by inhibiting tubulin polymerization because several different agents containing the quinolone ring structure have demonstrated antitubulin activity. COMPARE analysis corroborates the similarities between the anticancer agents identified here and various different microtubule inhibitors. With the exception of compound 319428, all of our compounds show strong similarity with colchicine, maytansine, vinblastine and vincristine. None of our compounds show significant relationship to taxol, which acts by stabilizing microtubules.
against the most tumorigenic of the NCI60 cell lines, based on the take rate of the cell lines in a mouse xenograft model. We also established that the activity of these compounds was not correlated to the expression of cell surface stem cell markers reported in the literature. Nevertheless, tumorigenic potential is the most important functional relationship between the most aggressive tumor cells and in vitro model for drug screening. Therefore, the anticancer agents identified based on their activity against the most tumorigenic cell lines may be considered as candidate anticancer agents that are specifically directed against subpopulations of cancer cells that drive the growth of tumors. One of these agents has been found to inhibit microtubule polymerization. Likewise, isosteres of three of our agents have also been shown to inhibit microtubule polymerization, suggesting a single mechanism of action. Interestingly, Compound 5388755 is structurally related to the potent antitubulin agent Combretastatin A-4. It is also possible that compound 379512 acts by inhibiting tubulin polymerization because several different agents containing the quinolone ring structure have demonstrated antitubulin activity. COMPARE analysis corroborates the similarities between the anticancer agents identified here and various different microtubule inhibitors. With the exception of compound 319428, all of our compounds show strong similarity with colchicine, maytansine, vinblastine and vincristine. None of our compounds show significant relationship to taxol, which acts by stabilizing microtubules.
Different inhibitors induce responses at distinct subsets of genes suggests that their functional effects
Rather than via their shared ability to induce histone hyperacetylation. Subsequent analysis of histone modification distributions on VPA-responsive genes showed that, irrespective of transcriptional response, histone acetylation at gene promoters does not reflect the inhibitor-induced increase in bulk histone hyperacetylation. Even after longer inhibitor treatments, of the eight genes examined only one, DLK1, showed a modest increase in H4 acetylation. This is a small sample, but includes all the genes that show the largest transcriptional responses to HDACi treatment, suggesting that our findings are representative of most genes. It seems that local levels of histone acetylation are determined by gene specific factors rather than induced changes in global histone modification. While there are examples of individual promoters that fail to show enhanced acetylation in response to HDACi, they have been seen as counterintuitive exceptions, and most reports focus on increased histone acetylation in response to HDACi. This remains controversial, as a recent study found HDACi induced transient increases in promoter acetylation at a subset of genes, but deacetylation after prolonged exposure was a more typical response. Our data is consistent with this, and BAY-60-7550 suggests that many promoters show minimal change in response to HDACi and that genes showing increased acetylation, such as the Hoxb genes are the exception rather than the rule. Signal Transducer and Activator of Transcription 3 belong to the STAT family of transcription factors. Compelling evidence has now established that aberrant STAT3 is a molecular abnormality that has a critical role in the development and progression of not only adult but also some pediatric tumors. In addition to its diverse biological functions including roles in cell proliferation, differentiation, apoptosis, inflammation, and oncogenesis, accumulating evidence suggests that STAT3 also plays an important role in cancer angiogenesis under both physiological and pathological situations. There is accumulating evidence that STAT3 is an important facilitator of tumor angiogenesis and its activation correlates with VEGF production in a variety of human cancers. In addition to its effects on VEGF, STAT3 has been implicated as a facilitator of angiogenesis by other mechanisms. For example, it has recently been demonstrated that STAT3 regulates expression of both MMP-2 and MMP-9, important facilitators of both angiogenesis and metastasis. It has been reported also that STAT3 is required for endothelial cell migration and microvascular tube formation. LLL12 is a novel small molecule allosteric inhibitor of STAT3, thought to bind STAT3 monomers at the tyrosine 705phosphorylation site and to prevent dimerization and activation. Previous work has established that LLL12 inhibits proliferation of various cancer cells in vitro, and tumor growth of both breast and glioblastoma xenograft models. Moreover, LLL12 induces apoptosis in medulloblastoma and glioblastoma cells and was also able to inhibit colony formation, wound healing and decreased IL6 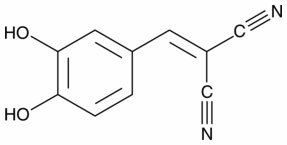 and LIF secretion. Antisense STAT3 oligonucleotide or STAT3 inhibitors, other than LLL12, have been shown to reduce microvessel density in tumor models. However, the mechanism for these anti-angiogenic effects has not been investigated. Our current work shows that at concentrations of drug that abrogate STAT3 phosphorylation, LLL12 blocks angiogenesis, and suppresses tumor vasculature in osteosarcoma tumors. The Adriamycin direct effect of LLL12 suppressing proliferation of HIVEC and HASMCs was shown at low concentrations of drug that completely suppressed VEGF-stimulation of STAT3 phosphorylation.
and LIF secretion. Antisense STAT3 oligonucleotide or STAT3 inhibitors, other than LLL12, have been shown to reduce microvessel density in tumor models. However, the mechanism for these anti-angiogenic effects has not been investigated. Our current work shows that at concentrations of drug that abrogate STAT3 phosphorylation, LLL12 blocks angiogenesis, and suppresses tumor vasculature in osteosarcoma tumors. The Adriamycin direct effect of LLL12 suppressing proliferation of HIVEC and HASMCs was shown at low concentrations of drug that completely suppressed VEGF-stimulation of STAT3 phosphorylation.