Recently, Zhang et al. reported twenty-five adult patients with HLH possessed genetic mutations in North America and Sieni et al. described eleven adult FLH in Italy. Various mutations in at least 9 genes have been shown to play roles in primary HLH. PRF1, UNC13D, STX11, (R)-(-)-Modafinic acid STXBP2, RAB27A, LYST, and AP3B1 are autosomal genes, all involving perforin/granzyme-mediated cytotoxicity. SH2D1A and BIRC4 are X-linked genes, coding for an adaptor molecule called signaling lymphocyte activation molecule-associated protein which is involved in signaling pathways that trigger cytotoxic granule release, and more recently, X-linked inhibitor of apoptosis. More importantly, all cases of HLH, regardless of genetic involvement, require prompt diagnosis and initiation of treatment. The aims of this study were to characterize genetic changes in a large population of adolescents and adults with HLH. The Beijing Friendship Hospital, Capital Medical University accepts specimens and referral cases from all over the country and has the largest adult HLH patient database in mainland China. For this study, we sequenced all coding exons and at least 50 base pairs of the adjacent intronic sequence of the PRF1, UNC13D, STX11, STXBP2, SH2D1A and BIRC4 genes in 252 adolescent and adult HLH patients from mainland China. We also measured NK cell activity to see if it correlated with certain mutations. Primary HLH was first proposed by Farquhar in 1952, who described high fever, pancytopenia, hepatosplenomegaly, and rapid death in 2-month-old twins. In subsequent reports, almost all patients were infants and young children, and more than half of the patients were twins, suggesting a genetic basis for the disease. Until a perforin gene mutation was found in a case of familial HLH, the diagnosis of primary HLH required onset in infancy and a positive family history as the support basis. After several additional genes and mutations were found to be associated with HLH, the Histiocyte Society developed 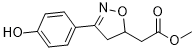 the diagnostic guidelines HLH-2004. Evidence of a genetic defect is one option for diagnosing primary HLH; however, patients with refractory/ reactivated HLH should also be considered as patients with severe disease. Adult HLH patients are typically diagnosed with secondary HLH because the disease seems to be induced by a concomitant condition, such as EBV infection, malignancy, or rheumatologic disorders. However, many of the genetic changes found in the pediatric primary HLH cases are also reported in adults. Here, we report genetic findings from a study of 252 adolescent and adult onset HLH patients from mainland China. Mutations in PRF1 occur in 15% to 50% of patients with primary HLH. Similarly, we found a PRF1 mutation in 9 of 18 patients in whom a mutation was identified. PRF1 is located at 10q21�C22 and has 3 exons, with all coding sequences in exons 2 and 3. It has been clearly documented that PRF1 mutations cause decreased or absent perforin protein expression on the surface of cytotoxic cells. This condition prohibits cytotoxic and NK cells from destroying their target cells, which in turn leads to increased cytokine production and macrophage activation, causing the symptoms of HLH. Cases of primary HLH with PRF1 mutations have been reported worldwide. At least 70 different mutations have been described between the two coding exons of PRF1, with p.W374X being the most common. Geographical and racial distributions of various PRF1 mutations have been reported. It is possible that the different PRF1 mutations Mycophenolic acid influence clinical severity.
the diagnostic guidelines HLH-2004. Evidence of a genetic defect is one option for diagnosing primary HLH; however, patients with refractory/ reactivated HLH should also be considered as patients with severe disease. Adult HLH patients are typically diagnosed with secondary HLH because the disease seems to be induced by a concomitant condition, such as EBV infection, malignancy, or rheumatologic disorders. However, many of the genetic changes found in the pediatric primary HLH cases are also reported in adults. Here, we report genetic findings from a study of 252 adolescent and adult onset HLH patients from mainland China. Mutations in PRF1 occur in 15% to 50% of patients with primary HLH. Similarly, we found a PRF1 mutation in 9 of 18 patients in whom a mutation was identified. PRF1 is located at 10q21�C22 and has 3 exons, with all coding sequences in exons 2 and 3. It has been clearly documented that PRF1 mutations cause decreased or absent perforin protein expression on the surface of cytotoxic cells. This condition prohibits cytotoxic and NK cells from destroying their target cells, which in turn leads to increased cytokine production and macrophage activation, causing the symptoms of HLH. Cases of primary HLH with PRF1 mutations have been reported worldwide. At least 70 different mutations have been described between the two coding exons of PRF1, with p.W374X being the most common. Geographical and racial distributions of various PRF1 mutations have been reported. It is possible that the different PRF1 mutations Mycophenolic acid influence clinical severity.