Physicochemical properties of binding of the peptides to core were measured by Fluorescence Polarization Light analysis, and by Surface Plasmon Resonance characterization of binding to mature core. Drug-like small molecules, identified using the assays developed to characterize the core-derived peptide inhibitors, displayed half-maximal inhibition of core dimerization and HCV infectivity at 90 nM concentrations. However, evidence for direct binding to HCV core protein in cells has lacked so far. We show here that a biotinylated derivative of SL209, one of these small molecule inhibitors, directly binds to HCV core presumably at the site of viral assembly in infected cells. 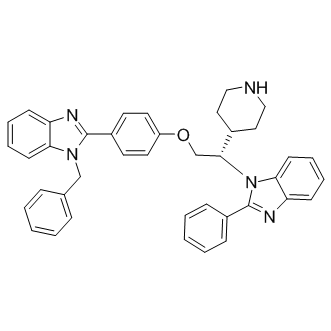 Ligandbased affinity isolation performed on lysates of HCV-infected cells or on recombinant HCV proteins demonstrated that the presence of core is required to retain other HCV proteins on the affinity-gel, thus confirming the central role of core in virion assembly. We describe here the first evidence of binding, to the HCV capsid protein, of a core dimerization inhibitor which reduces HCV production and infectivity. Direct binding was shown by using a biotinylated derivative of small molecule drug-like SL209, that largely maintained the HCV inhibitory properties of the untagged compound. Using SL209-biotin absorbed on agarose beads coated with streptavidin, direct physical interaction was demonstrated by affinity-isolation performed on lysates of HCVinfected cells, and confirmed with recombinant HCV proteins. Affinity-binding was shown not only for core, but also for other HCV proteins which were previously reported to be co-localized with core on lipid droplets or on ER, namely NS3, NS5A, and NS5B. In the absence of core, neither NS5A nor NS3 were retained on the SL209-biotin coated streptavidin beads. These results confirm that in the affinity-isolation conditions, SL209-biotin binding is strong enough to retain core complexes containing other HCV proteins. In support of this observation, colocalization of core and NS5A proteins with SL209-biotin was occasionally observed in HCV-infected cells using ICG-001 confocal microscopy. Despite similar structures, SL209-biotin may thus differ significantly from another inhibitor SL201, which had earlier been shown to not only disrupt core dimerization, but also inhibit interaction with NS3 helicase. Alternatively, assay conditions and their effect on avidity and affinity may play a major role in SCH772984 dictating complex formation: in cells, or on agarose beads. Protein-ligand complexes may be more stable than in the transferof-energy assays optimized to measure inhibition of proteinprotein interactions. Using SL209-biotin in combination with Streptavidin-Alexa 488, we showed by confocal immunofluorescence microscopy that the presence of core is indispensable to observe any intracellular staining: no labeling is observed in uninfected cells nor in cells infected with HCV replicon or with HIV. Furthermore, SL209biotin staining coincides mostly, but not always, with immunostaining of core. Cellular co-staining of core and SL209-biotin occurs often in sites where we could not detect lipid droplets, mainly on Endoplasmic Reticulum, as determined by staining with LAMP1, an ER marker protein. This observation is in line with the recent discovery that the core protein of high-titer JC1 recombinant HCV virus, used in our studies frequently exhibits an ER localization, rather than the predominant lipid droplet localization of the core protein of JFH1 virus described in previous co-localization studies in HCV-infected cells and core-transfected uninfected cells. Our choice of using JC1 in preference to JFH1 was dictated by a much improved level of viral production.
Ligandbased affinity isolation performed on lysates of HCV-infected cells or on recombinant HCV proteins demonstrated that the presence of core is required to retain other HCV proteins on the affinity-gel, thus confirming the central role of core in virion assembly. We describe here the first evidence of binding, to the HCV capsid protein, of a core dimerization inhibitor which reduces HCV production and infectivity. Direct binding was shown by using a biotinylated derivative of small molecule drug-like SL209, that largely maintained the HCV inhibitory properties of the untagged compound. Using SL209-biotin absorbed on agarose beads coated with streptavidin, direct physical interaction was demonstrated by affinity-isolation performed on lysates of HCVinfected cells, and confirmed with recombinant HCV proteins. Affinity-binding was shown not only for core, but also for other HCV proteins which were previously reported to be co-localized with core on lipid droplets or on ER, namely NS3, NS5A, and NS5B. In the absence of core, neither NS5A nor NS3 were retained on the SL209-biotin coated streptavidin beads. These results confirm that in the affinity-isolation conditions, SL209-biotin binding is strong enough to retain core complexes containing other HCV proteins. In support of this observation, colocalization of core and NS5A proteins with SL209-biotin was occasionally observed in HCV-infected cells using ICG-001 confocal microscopy. Despite similar structures, SL209-biotin may thus differ significantly from another inhibitor SL201, which had earlier been shown to not only disrupt core dimerization, but also inhibit interaction with NS3 helicase. Alternatively, assay conditions and their effect on avidity and affinity may play a major role in SCH772984 dictating complex formation: in cells, or on agarose beads. Protein-ligand complexes may be more stable than in the transferof-energy assays optimized to measure inhibition of proteinprotein interactions. Using SL209-biotin in combination with Streptavidin-Alexa 488, we showed by confocal immunofluorescence microscopy that the presence of core is indispensable to observe any intracellular staining: no labeling is observed in uninfected cells nor in cells infected with HCV replicon or with HIV. Furthermore, SL209biotin staining coincides mostly, but not always, with immunostaining of core. Cellular co-staining of core and SL209-biotin occurs often in sites where we could not detect lipid droplets, mainly on Endoplasmic Reticulum, as determined by staining with LAMP1, an ER marker protein. This observation is in line with the recent discovery that the core protein of high-titer JC1 recombinant HCV virus, used in our studies frequently exhibits an ER localization, rather than the predominant lipid droplet localization of the core protein of JFH1 virus described in previous co-localization studies in HCV-infected cells and core-transfected uninfected cells. Our choice of using JC1 in preference to JFH1 was dictated by a much improved level of viral production.